Quantitative Analysis of Cancer-associated Gene Methylation Connected to Risk Factors in Korean Colorectal Cancer Patients
Article information
Abstract
Objectives
The purpose of this paper was to elucidate the potential methylation levels of adjacent normal and cancer tissues by comparing them with normal colorectal tissues, and to describe the correlations between the methylation and clinical parameters in Korean colorectal cancer (CRC) patients.
Methods
Hypermethylation profiles of nine genes (RASSF1, APC, p16INK4a, Twist1, E-cadherin, TIMP3, Smad4, COX2, and ABCB1) were examined with 100 sets of cancer tissues and 14 normal colorectal tissues. We determined the hypermethylation at a given level by a percent of methylation ratio value of 10 using quantitative methylation real-time polymerase chain reaction.
Results
Nine genes' hypermethylation levels in Korean CRC patient tissues were increased more higher than normal colorectal tissues. However, the amounts of p16INK4a and E-cadherin gene hypermethylation in normal and CRC tissues were not significantly different nor did TIMP3 gene hypermethylation in adjacent normal and cancer tissues differ significantly. The hypermethylation of TIMP3, E-cadherin, ABCB1, and COX2 genes among other genes were abundantly found in normal colorectal tissues. The hypermethylation of nine genes' methylation in cancer tissues was not significantly associated with any clinical parameters. In Cohen's kappa test, it was moderately observed that RASSF1 was related with E-cadherin, and Smad4 with ABCB1 and COX2.
Conclusions
This study provides evidence for different hypermethylation patterns of cancer-associated genes in normal and CRC tissues, which may serve as useful information on CRC cancer progression.
INTRODUCTION
Colorectal cancer (CRC) arises due to genetic alterations through gene mutations and methylation and transforms colorectal epithelial cells into colorectal adenocarcinoma cells [1]. CRC is suitable for early detection approaches because it is recognizable at early stages [2]. Early detection of CRC appears to be most important in reducing mortality [3]. Therefore, molecular studies on epigenetic changes including CRC instability and DNA methylation have both been investigated for better understanding of CRC progression [4,5].
DNA methylation effects of a number of mediating genes in CRC development and the significance of the aberrant methylation in CRC have been reported [3,6,7]. The hypermethylation of several promoters including APC, p16INK4a, and TIMP3 in CRC [8,9] and the methylation-induced silencing of cancer suppressor genes were investigated by a quantitative approach [10]. A quantitative approach toward methylation changes with normal and cancer tissues could provide useful information for a panel of DNA methylation markers for the detection of cancer occurrence.
In this study, we evaluate the potential methylation of cancer-associated genes concerned with cell cycle control, invasion, cancer progression, drug metabolism, and inflammation in Korean CRC tissues. Subsequently, we compare the methylation statuses of selected genes in CRC and fourteen samples of normal colorectal tissue and examine whether the amount of hypermethylation is associated with clinical parameters of CRC patients.
METHODS
Tissue Samples
A total of 100 paired CRC tissue samples containing adjacent normal tissue were obtained from CRC patients at Dong-A University Hospital in Busan, Korea. The diagnosis of CRC tissues was acquired from pathology reports and histological evaluations. Fourteen normal colorectal tissue samples were obtained from dead victims of traffic accidents who had agreed to body donation for medical research and education. Age and sex distributions were not significantly different among the subjects in the patient and normal groups. The design of this study was approved by the Committee on Human Research of Dong-A University. Every fresh tissue sample was frozen in liquid nitrogen right after the resection and storage in a deep refrigerator at -80℃.
Genomic DNA Extraction and Modification
Genomic DNA was extracted by means of a QIAamp DNA mini kit (Qiagen, Valencia, CA, USA). The tissue samples were ground with a pestle in liquid nitrogen and treated with 700 µL of a lysis buffer containing 20 µg/mL LaboPass protease K (Cosmo Gene Tech, Seoul, Korea), 20 mM Tris·HCl (pH 8.0), 5 mM EDTA (pH 8.0), 400 mM NaCl and 1% SDS solution (Sigma, St. Louis, MO, USA). The samples were then incubated in a 42℃ incubator overnight. After incubation, the genomic DNA was purified three times via phenol/chloroform extraction. It was then eluted with 300 µL of water. The DNAs were quantified with a NanoDrop ND-100 device (Thermo Fisher Scientific, Waltham, MA, USA).
DNA modification was performed by means of an EZ DNA modification kit (Zymo-Research, Orange, CA, USA) following the manufacturer's recommended procedure. One microgram of the extracted DNA was mixed with up to 50 µL of sodium-bisulfite containing 5 µL of M-dilution buffer. This mixture was then incubated for 15 minutes at 37℃ in an incubator. One hundred microliters of CT conversion reagent containing M-dilution buffer was added to each sample and this was incubated for 15 hours at 50℃ in an incubator without light. After this 15 hours incubation period, the DNA samples were placed on ice for 10 minutes. They were then transferred into Zymo-spin IC columns, treated with 400 µL of M-binding buffer, and then centrifuged at 13 000 rpm for 30 seconds. After discarding the flow-through material, 200 µL of M-wash buffer was added to the columns, and the resultant solution was centrifuged at 13 000 rpm for 30 seconds. After washing, 200 µL of M-desulphonation buffer was added to the samples. The samples were subsequently incubated at room temperature for 15 minutes and then centrifuged at 13 000 rpm for 30 seconds. Each column was washed two times with 200 µL of M-wash buffer. The modified genomic DNA was eluted using 30 µL of water.
Primer and Probes for Quantitative Methylation Reverse Transcription Polymerase Chain Reaction
We used previously reported primer and probe sequences for nine cancer associated genes [11-13]. These sequences are summarized in Table 1. Amplification of bisulfite-converted ACTB was used for the normalization of the input DNA. The negatives for the ACTB samples were excluded from the methylation analysis. A plasmid containing a bisulfite-converted ACTB gene of a known concentration was drawn at 1, 1/4, 1/16, 1/64, and 1/256 via serial dilutions. This was used as the standard curve for quantification.
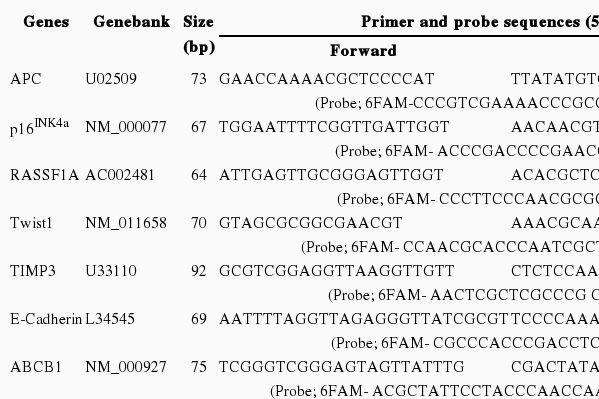
Primer and probe sequences for quantitative methylation reverse transcription polymerase chain reaction
Reverse transcription polymerase chain reaction (RT-PCR) was performed by TaqMan gene assays by means of a 7200 HT Fast real-time PCR system (Applied Biosystems, Foster City, CA, USA) and FAM-labeled probes (Table 1). Each PCR program started with an initial denaturation at 95℃ for 10 minutes, followed by 45 cycles of denaturation at 95℃ for 15 seconds, and annealing/extension at 60℃ for 1 minute. Sodium-bisulfite treated CpGenome universal methylated DNA (Chemicon, Temecula, CA, USA) was used as a positive control. Relative quantization of the amplified gene levels in the tested genomic DNA samples was made by measuring the threshold cycle (CT) values of the target genes and the beta-actin. The mean quantity of the genes was divided by the mean quantity of the ACTB. The RT-PCR reaction was duplicated.
Statistical Analysis
Hypermethylation was determined by the percentage of methylation ratio (PMR), and PMR values over 4% (PMR 4) or 10% (PMR 10) were utilized [14-17]. In our study, a PMR value was defined as ([gene] sample / [ACTB] sample) / ([gene] control / [ACTB] control)×100 and hypermethylation of each gene was defined by PMR 10. The significance of the differences in the methylation ratio (i.e., PMR) and hypermethylation frequencies among normal tissues, adjacent normal tissues, and cancer tissues was evaluated with a t-test, chi-squared test, and Fisher's exact test using SPSS version 12.0 (SPSS Inc., Chicago, IL, USA). All of the statistical tests were two-sided, and the statistical significance was deemed acceptable at p<0.05. Cohen's kappa statistic was used to measure the agreement among the methylation of nine gene promoters (PMR cut-off value of 10). The kappa values were interpreted as follows: <0.2, poor observer agreement; 0.2-0.4, fair observer agreement; 0.4-0.6, moderate observer agreement; 0.6-0.8, substantial observer agreement; and 0.8-1.0, good observer agreement [18].
RESULTS
We performed the statistical analysis by defining the proportion of hypermethylation as a given PMR value of 10 (PMR 10). The hypermethylation of nine cancer-associated genes showed an increase in the adjacent normal and cancer tissues among CRC patients compared with normal colorectal tissues (Figure 1). The hypermethylation of nine genes for both adjacent normal and cancer tissues in CRC patients was higher more increased than that for normal tissues. However, the amounts of p16INK4a and E-cadherin gene hypermethylation in normal and cancer tissues were not significantly different and TIMP3 gene hypermethylation was not significantly different in adjacent normal and cancer tissues (Figure 1). The average PMR values and hypermethylation frequencies from the quantitative methylation RT-PCR are summarized in Table 2. Among the nine cancer-associated genes, hypermethylation of TIMP3, E-cadherin, ABCB1, and COX2 genes were abundantly found in normal colorectal tissues within the PMR 10. However, the average PMR values of each gene other than E-cadherin were higher in the cancer tissues than in the normal tissues (Table 2). The p16INK4a gene hypermethylation frequencies for the adjacent normal and cancer tissues with PMR 10 were 10% and 14%, respectively. No significant difference was revealed (Table 2) (p>0.05). The association between hypermethylation and clinical parameters in the 100 CRC patients in accordance with PMR 10 is presented in Table 3. The hypermethylation of nine cancer-associated genes in CRC tissues was not associated with any clinical parameters including age, gender, smoking status, alcohol intake, or cancer staging. With Cohen's kappa test, it was observed that RASSF1 was moderately related to E-cadherin, and Smad4 with ABCB1 and COX2 (Table 4).
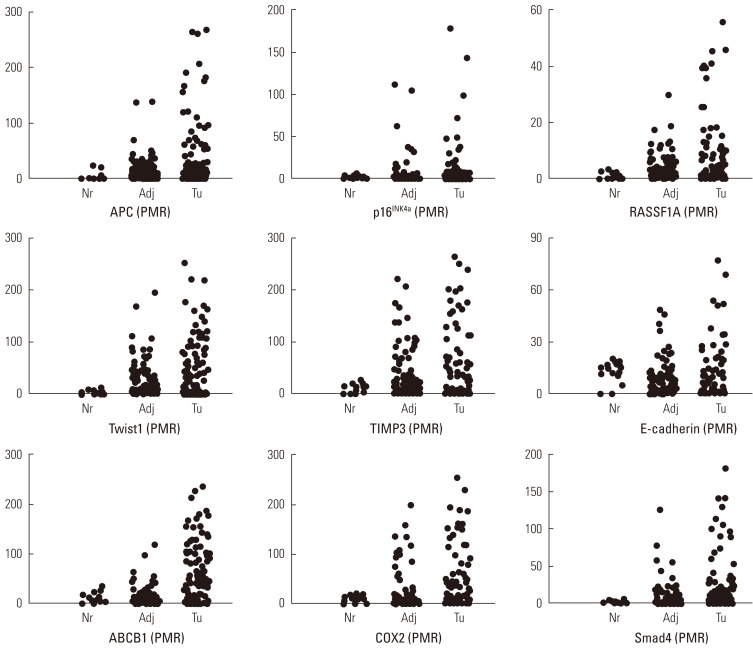
Percentage of methylation ratio (PMR) contains nine cancer-associated genes in colorectal cancer patients. Promoter methylation of nine tumor-association genes in tumor tissues and corresponding adjacent normal tissues from 100 patients with colorectal cancers and fourteen normal colorectal tissues was measured by means of quantitative methylation reverse transcription polymerase chain reaction. Nr, normal colorectal tissues; Adj, adjacent normal colorectal tissues; Tu, colorectal cancer tissues.
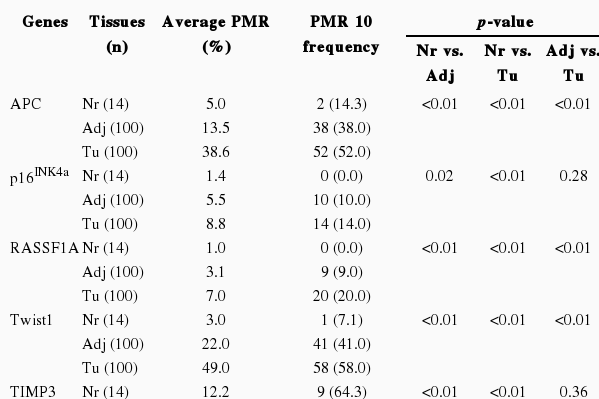
Average PMR and hypermethylation frequency over PMR 10 according to gene and tissue type
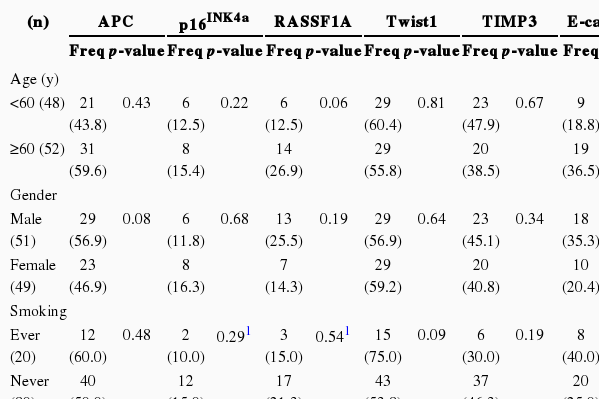
Hypermethylation frequencies over PMR 10 of nine cancer-associated genes with clinical parameters in 100 colorectal cancer tissues

Hypermethylation agreement (kappa values) among nine cancer-associated genes in 100 colorectal cancer tissues
DISCUSSION
In the current study, the main objective was to elucidate the potential methylation levels of adjacent normal and cancer tissues by comparing them with normal colorectal tissues in Korean CRC patients. In addition, we sought DNA methylation profiles and their association with clinical parameters in CRC. The hypermethylation of eight cancer-associated genes, that is, all except for the E-cadherin gene, was higher in adjacent normal and cancer tissues than that in normal colorectal tissues, suggesting a fundamental role in the methylation of cancer-associated genes in Korean CRC patients.
The APC gene mechanisms are associated with gene inactivation by promoter hypermethylation, and may also play a role in the loss of APC function in tumors [6,19]. The promoter region of the APC gene is highly methylated in colorectal carcinoma compared to normal colorectal mucosa and premalignant adenomas [20]. In addition, Rashid et al. [7] reported that methylation was more frequent in adenomas with tubulo-villous or villous histology. APC hypermethylation has been also considered an early step in the process of CRC pathogenesis and cross-talk with the WNT/β-catenin pathway [8,21]. Likewise, in the present study, an APC gene methylation state was observed in CRC tissues and adjacent normal tissues. These results may be supported by the finding that APC hypermethylation may ultimately lead to tumor development [22]. Cyclooxygenase2 (COX2 or PTGS2) was also engaged in cross-talk with the WNT/β-catenin pathway [23]. Castells et al. [24] have reported that the hypermethylation of COX2 appears to be the most attractive explanation for the lack of COX2 overexpression in cancer patients. In addition, COX2 promoter hypermethylation was significantly associated with a lack of COX2 expression. We have determined that COX2 hypermethylation may support regulatory mechanisms in CRC. Thus, the evidence suggests that the COX2 hypermethylation mechanism is involved in the response to COX2 gene silencing. In our additional data, the APC and COX2 promoter methylation in CRC tissues was not significantly associated with any clinical parameters such as age, gender, or smoking status according to PMR 10. Lin et al. [25] showed that APC promoter methylation was significantly higher in cancerous tissues than non-cancerous tissues, but the methylation status had no clear relationship with clinical parameters. APC and COX2 methylation studies are needed to confirm the findings on the association with clinical parameters by a sufficient number of subjects.
Previous studies have reported the aberrant methylated incidence associated with inactivation of p16INK4a in CRC [18,26]. Wiencke et al. [27] reported that methylation of p16INK4a was associated with anatomical location, gender, age, and poorly differentiated adenocarcinoma. Ishiguro et al. [28] also observed the methylation of p16INK4a in 20/88 (22.7%) of cancer tissues, but the frequency of p16INK4a methylation in cancer tissues did not show a significant difference from that in normal mucosae. In addition, associations between methylation of p16INK4a and clinical features such as age and cancer staging have been identified. Our results also found 14% p16INK4a gene hypermethylation in cancer tissues but none in normal tissues; however, p16INK4a methylation in cancer tissues was not significantly different from adjacent normal tissues. In addition, p16INK4a methylation in cancer tissues was not significantly associated with any clinical parameters. Although p16INK4a hypermethylation in cancer tissues in CRC patients seems to show a few differences among ethnicities, it remains a promising biomarker for CRC.
Wagner et al. [29] investigated RASSF1A promoter methylation in CRC and detected RASSF1A methylation in 80% (4/5) of CRC cell lines and 45% (13/29) of primary CRCs. Xu et al. [30] found that the frequency of RASSF1A methylation was extremely low in CRC. In our data, a similar low methylation frequency in the RASSF1A gene was observed in 20% of cancer tissues and 9% of adjacent normal tissues. However, in comparing cancer tissues and normal tissues, RASSF1A gene methylation showed significant differences. Also, the RASSF1A methylation in cancer tissues was 2-fold higher among the subjects over age 60. Age-related epigenetic defects have been proposed as potential sources of the field defects in colorectal carcinogenesis [31]. These data support the possibility that age-related RASSF1A methylation is involved in the pathways for the promotion of cancer progression of CRCs.
The methylation and subsequent loss of cell-cell adhesion may favor cancer progression and metastasis. Increasing hypermethylation of cell adhesion genes such as Twist1 and TIMP3 was observed in both adjacent normal and cancer tissues in our study. Therefore, the hypermethylation of these genes may play a potentially crucial role in CRC progression, most likely promoting cancer invasion. The presence of hypermethylation in adjacent normal tissues obtained from patients suggests that it might be an early event in the process of colorectal carcinogenesis. However, E-cadherin gene methylation frequency was not significantly different between cancer and normal colorectal tissues. Twist1 hypermethylation was more frequently found in cancer tissues than in their paired non-neoplastic tissues, suggesting that Twist1 might be considered a Type-C (cancer-related CpG methylation) agent with the potential to be used as an epigenetic cancer biomarker [32]. In other reported data, Twist1 hypermethylation was observed in colorectal specimens of 46 patients with cancer, but in none of the tissues from a nonmalignant control group [33]. Okada et al. [34] observed a higher Twist1 methylation level in colorectal adenoma and cancer tissues than in normal colorectal mucosa. However, there was no correlation between Twist1 methylation and expression. These results suggest the possibility that Twist1 methylation may not influence the promotion of CRC invasion by alternative compensatory molecular pathways, but the association of these findings with any clinical parameters must be confirmed. Aberrant expression of ABCB1 (MDR1) is associated with various carcinomas. The hypermethylation of normal CpG islands that were located in the promoter regions involved gene silencing at the transcriptional level [35]. We observed an increasing rate of ABCB1 hypermethylation in both adjacent normal and cancer tissues of CRC patients. The results suggest that ABCB1 hypermethylation is a candidate biomarker for CRC progression. However, ABCB1 promoter methylation in cancer tissues was not significantly associated with any clinical parameters.
TGF-β overexpression and Smad4 mutations or deletions have been directly correlated with CRC metastasis [36]. In the current study, the methylation of the Smad4 gene was observed in CRC tissues and adjacent normal tissues. Our finding that Smad4 hypermethylation occurred in CRC may support an association with cancer metastasis, which may ultimately lead to cancer development. However, Smad4 promoter methylation in CRC tissues was not significantly associated with any clinical parameters.
In conclusion, the key strength of the current research is that normal human tissues and CRC tissues are examined in an epigenetic study. The results of this study provide evidence that hypermethylation of cancer-associated genes may possibly serve as useful information regarding CRC cancer progression. Due to its small numbers, the current study has limitations as an association study; therefore, further studies with a sufficient number of subjects and additional candidate genes are required to confirm these findings.
ACKNOWLEDGEMENTS
This paper was supported by the Dong-A University Research Fund.
Notes
The authors have no conflicts of interest with the material presented in this paper.